
Simulating a virtual cell with all its underlying bioprocesses is one of the holy grails of medicine. While Markus Covert’s group from Stanford has published a whole-cell simulation of mycoplasma capturing more than 20 bioprocesses at a mesoscale level, the models comprising the simulation was built entirely by hand, and are not data-driven. This means that additional data cannot be easily used to refine it further. In addition, the mesoscale level does not allow modelling subtle genetic mutations, or the refinement of 3D structure of drugs that can induce changes in the cell.
More recently, a structural model of a cell was recently published by Maritan et al: https://ccsb.scripps.edu/gallery/mycoplasma_model/.
We've been building a #molstar app for exploring mesoscale models with Billions of atoms. If you dare, try the examples in the Mesoscale Explorer — BETA, may 🔥🖥️ https://t.co/lmWcQ5HCeq@sehnaldavid @grinche pic.twitter.com/pnyYTUhdzu
— Alexander Rose (@asrmoin) November 6, 2023
This type of model suggests that a first-principles approach called Molecular Dynamics can be potentially used to simulate it. Molecular Dynamics is a computational method for simulating the movement of atoms and molecules. For each atom in a given protein, several forces are computed and then integrated in time to estimate the dynamic motion of the atoms and of molecules. The movements of atoms are computed by calculating forces at each timestep for each atom, and then integrating those forces over time, thus solving a differential equation.
While a virtual cell is not possible to meaningfully simulate at the moment using Molecular Dynamics, a more tractable target is to simulate the formation of virus capsids through self-assembly. Virus capsids (capsules containing the viral DNA) is often made of 60,90,120 or more proteins that attract each other to form a highly-symmetric capsule called a capsid:
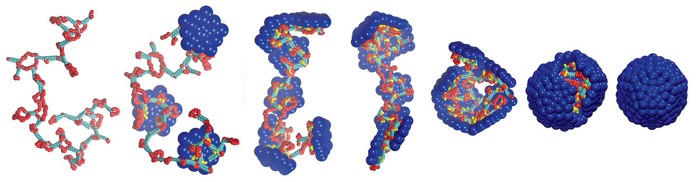
Even virus self-assembly is difficult. Molecular dynamics simulations are computationally very intensive, and the time required to achieve a complete assembly of a virus capsid using traditional MD would be astronomical (decades to centuries of GPU time). Therefore, we are using several statistical techniques such as enhanced sampling and ML-derived potentials to achieve speed-ups in the order of 10,000X or more. For speeding up MD simulations with ML, see our other project.
I’m a student interested in working on this. What do I do?
If you’ve never heard of Molecular Dynamics, run 1-2 simulations in OpenMM first using some protein structure downloaded from the protein data bank (RCSB). Google for “OpenMM tutorial/getting started”. Once you generate a trajectory, try to visualize it using nglviewer. You should see the atoms moving around and the protein “wriggle”, which is an expected behaviour given the short simulation you’ve just run. Then look through the further reading below and try to understand the papers. Once you are decided that you’d like to work on this project, send me an email.
Further reading:
- Molecular dynamics simulations of an entire cell: https://www.frontiersin.org/articles/10.3389/fchem.2023.1106495/full
- Building Structural Models of a Whole Mycoplasma Cell: https://pubmed.ncbi.nlm.nih.gov/34774566/
- Multi-scale models of whole cells: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10661945/
- Mechanisms of Virus Assembly: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4382372/
- Multiscale modeling of hepatitis B virus capsid assembly and its dimorphism: https://pubs.acs.org/doi/pdf/10.1021/acsnano.2c02119
- 1-billion atom COVID simulation: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8609898/
Enhanced sampling:
- WESTPA paper (technique for accelerating rare events by factors up to 1,000x): https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4573570/
- WESTPA 2.0 tutorials: https://livecomsjournal.org/index.php/livecoms/article/view/v5i1e1655
- Umbrella sampling: https://en.wikipedia.org/wiki/Umbrella_sampling
- Metadynamics: https://wires.onlinelibrary.wiley.com/doi/pdf/10.1002/wcms.31
- Parallel tempering: https://en.wikipedia.org/wiki/Parallel_tempering